

De la historia clínica electrónica al diagnóstico genético
El fenotipo de algunas enfermedades puede ser complejo y presentar un evolutivo multidimensional. Estas diferencias suelen ser capturadas por las imágenes diagnósticas, diferentes análisis bioquímicos, y el uso de biomarcadores de precisión, ensayos que se pueden ser solicitados en diferentes momentos de la enfermedad y por diversos profesionales en el curso de la identificación de un diagnóstico diferencial (21). Estos resultados se documentan en la historia clínica electrónica donde se sintetizan los hallazgos para facilitar la toma de decisiones y comunicar las decisiones clínicas. Los sistemas de IA facilitan el reconocimiento de patrones en estos registros. Un estudio reciente que involucró más de 500.000 pacientes utilizó un modelo para procesamiento natural del lenguaje (Natural language processing) basado en IA para extraer características clínicamente relevantes de la información consignada (22). Después de entrenar al modelo estadístico jerárquico y escalonado sobre la base de divisiones anatómicas se generó un sistema diagnóstico adaptado capaz de diferenciar 55 diagnósticos pediátricos comunes con un 92% de precisión. En paralelo, el uso del modelo vinculado a información genómica permitió estratificar enfermedades raras en los niños y clasificar coincidencias con variantes patogénicas extraídas del análisis del genoma de los pacientes (23). En 101 niños con 105 enfermedades genéticas, la valoración retrospectiva de los diagnósticos genómicos por vía automatizada coincidió con la interpretación humana experta con un 99% de precisión (24).
Desafíos en la interpretación de la genómica tumoral
La secuenciación de próxima generación (NGS, Next Generation Sequencing) ha revolucionado la investigación biomédica permitiendo la generación de estudios multicapa que integran datos genómicos en diversas dimensiones, incluyendo el ADN-seq y la ARN-seq, así como información multiómica que incluye al proteoma, epigenoma, metaboloma, microbioma, etc (25). Esta fusión proporciona una visión más completa de los procesos y sistemas biológicos, lo que conduce a una mejor comprensión de la enfermedad, especialmente en comparación con el análisis de una sola capa. Sin embargo, existen varios desafíos para la traducción de datos multiómicos en biomarcadores clínicamente procesables. Primero, combinar perfiles de datos en varios niveles daría como resultado una alta dimensionalidad con un gran número de covariables. La escasez de datos de alta dimensionalidad asociada a la alta heterogeneidad entre los diversos tipos de datos impone una dificultad significativa en los análisis integrativos. Se han desarrollado diferentes técnicas para reducir estas dimensiones, incluyendo el análisis de co-inercia múltiple y el análisis de factores múltiples, diseños útiles para facilitar los análisis en la transcripción corriente abajo (down stream signaling). Varios marcos se han utilizado para la integración, incluyendo los enfoques basados en redes que utilizan algoritmos gráficos para capturar interacciones potenciales entre las redes moleculares, y modelos bayesianos de varios niveles que imponen suposiciones realistas para la estimación de parámetros a través de una estructura de entrada y salida (26,27). En segundo lugar, la integración de datos multiómicos requiere la mejor progresiva de los estándares para la generación de resultados, facilitando la interpretación y reduciendo el sesgo.
Por otra parte, los procedimientos para la adquisición y preparación de las muestras deben estar correctamente regulados para cada una de las plataformas de secuenciación y entre ellas. Por ejemplo, para la información derivada del análisis por NGS se necesita material de referencia (CLSI QMS01-A 2018; CLSI MM01A3E 2018; NIST 2018) cuyas propiedades sean suficientemente homogéneas y estén bien establecidas para la calibración del sistema de secuenciación. Por último, pero no menos importante, se necesitan estudios bien diseñados que permitan hacer inferencia causal para filtrar los biomarcadores que tienen fuertes efectos predictivos (28).
La diversidad de la evidencia puede contribuir con la inferencia patogénica de las variantes, incluidos datos genéticos, informáticos y experimentales. A nivel genético, las variantes patogénicas pueden enriquecerse significativamente a partir del análisis de casos y controles y/o ante la evidencia de una variante germinal que afecta el estado de la enfermedad dentro de una familia afecta. En el nivel informático, las variantes patogénicas se pueden encontrar en el lugar que se predice que causará una alteración funcional (región de unión a proteínas). Y a nivel experimental, las variantes patogénicas pueden alterar significativamente los niveles, el empalme o la función bioquímica normal del producto de los genes afectados. Esto puede mostrarse en células de pacientes o bien puede ser validado con modelos in vitro o in vivo (29,30).
El avance de las tecnologías relacionadas con machine learning está destinado a afectar la interpretación de los datos provenientes de la secuenciación genómica, que tradicionalmente se basó en la curación manual. Estos esfuerzos de purificación se basan en la estructura de proteínas, estudios funcionales y, más recientemente, en modelos “in silico” que predicen el impacto funcional de la alteración genética usando plataformas como SIFT, PANTHER-PSEP, PolyPhen2 y otros (30). En adición, las bases de datos genómicas como ClinVar, COSMIC y OncoKB han proliferado como medio para compilar de manera concisa una colección de las variantes genéticas (Figura 3). En general, proporcionan la evidencia que respalda la clasificación de una variante como patogénica, benigna o de significado desconocido (VUS).
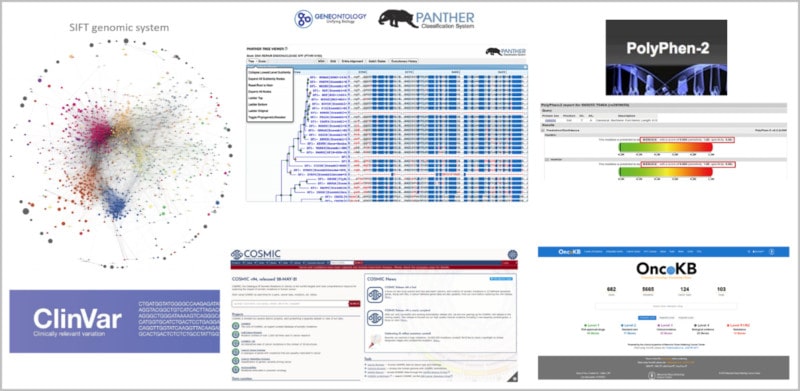
Figura 3. Repertorio de plataformas que integran IA para el llamado y análisis de variantes génicas.
Dos de las limitaciones clave de la curación e interpretación manual de los resultados derivados de datos genómicos crudos son la escalabilidad y la reproducibilidad. Estos desafíos continúan creciendo a medida que se dispone de más información. La cantidad de expertos en clasificación de variantes y la cantidad de tiempo que pueden dedicar diariamente a esta tarea es limitada. Para abordar estas limitaciones, varias organizaciones están trabajando en crear y estandarizar protocolos para la clasificación de variantes, incluyendo el American College of Medical Genetics and Genomics y la Association for Molecular Pathology (ACMG-AMP), quienes ya publicaron una serie de directrices para la interpretación de variantes genéticas de la línea germinal y somática para genes causantes de trastornos hereditarios y del cáncer (31,32). Sin embargo, la capacidad de escalar la interpretación de variantes provenientes de los estudios de NGS, especialmente en cáncer, sigue siendo limitada, requiere validación y un estricto control de calidad (33). Recientemente se presentó la plataforma OncoTree que incluye 886 tipos de tumores originados en 32 complejos tisulares; esta plataforma fue adoptada como sistema de clasificación para el proyecto Genomics Evidence Neoplasia Information Exchange (GENIE) de la Asociación Estadounidense para la Investigación del Cáncer (AACR), un gran consorcio de intercambio de datos genómicos y clínicos, para amplificar y unificar el esfuerzo de OncoKB y cBioPortal for Cancer Genomics (34).
Cómo la integración de la NGS y la IA están cambiando el panorama de la genómica tumoral
Actualmente, la NGS se aplica ampliamente como método valioso para obtener un perfilamiento genómico exhaustivo. Gracias a esta tecnología se ha logrado secuenciar simultáneamente millones de fragmentos de ADN en una sola muestra para detectar una amplia gama de aberraciones propias del cáncer. Los paneles de cáncer están diseñados específicamente para detectar mutaciones somáticas y germinales clínicamente relevantes. De igual forma, la caracterización molecular usando NGS por biopsia líquida facilita el diagnóstico temprano, la evaluación de la heterogeneidad tumoral y de la enfermedad mínimas residual siguiendo un principio no invasivo. Gracias a estos modelos de tipificación genómica se abrieron proyectos como el Cancer Genome Atlas (TCGA) que ha permitido el descubrimiento de nuevos mecanismos oncogénicos y la estratificación de pacientes y enfermedades (35). La información utilizada para dilucidar los mecanismos primarios en la evolución del cáncer ha permitido elucidar el metabolismo oxidativo de las células tumorales (36), validar la utilidad de biomarcadores predictivos como la metilación del promotor de la MGMT en tumores de estirpe glial (37), proyectar el efecto terapéutico de la inmunoterapia en cáncer gástrico (38), confirmar la utilidad in silico de mutaciones particulares en cáncer de pulmón (38), y considerar a la transición epitelio mesenquimal como parte de la resistencia en cáncer de seno (39), entre muchos otros.
Uno de los mayores retos de la genómica tumoral está asociado con el llamado, depuración e interpretación de las variantes. Frecuentemente, los usuarios necesitan ajustar los parámetros de forma heurística y aplicar filtros personalizados para eliminar los falsos positivos antes de lograr una precisión aceptable. Este es un esfuerzo que requiere tiempo y experiencia para ajustar las puntuaciones de calidad y los atributos dentro de los contextos de secuenciación. Diferentes grupos están aprovechando algoritmos de aprendizaje automático y el entrenamiento en las características de calidad subyacentes para mejorar el rendimiento del llamado de variantes, especialmente en escenarios subóptimos (40-43). Estos, han permitido establecer el valor de la ploidía como factor que contribuye con la complejidad de la enfermedad. De igual forma, contribuyeron para establecer el valor de las variantes subclonales (presentes solo en unas pocas células), difíciles de detectar porque su representación en la librería de secuenciación suele ser baja. Este hallazgo resulta en una mayor variabilidad entre los métodos de análisis, los umbrales y las puntuaciones de calidad que pueden no ser lo suficientemente flexibles para detectar la evolución subclonal de la enfermedad (41). En lugar de configurar reglas estáticas, los métodos de IA permiten ajustar los umbrales de forma dinámica en función de los patrones de expresión génica. Las variantes con frecuencias alélicas muy bajas aún se pueden informar si la profundidad de secuenciación y otras métricas de calidad superan los umbrales. Por ejemplo, un modelo de red neuronal convolucional cuyos algoritmos se utilizan a menudo en el reconocimiento de imágenes logró una puntuación F1 de 0.96 y pudo alcanzar variantes con una frecuencia de alelos tan baja como 0.0001 (la puntuación F1 es una medida que tiene en cuenta tanto la precisión como la memoria) (44). En otro caso, un enfoque basado en machine learning aplicado a los datos de NGS mostró una precisión mejorada (medida por la puntuación F1) en la identificación de mutaciones tumorales en comparación con otros programas existentes como MuTect1, MuTect2, SomaticSniper, Strelka, VarDict y VarScan2. Si bien sus valores de recuperación fueron similares, la plataforma de IA mostró mayor precisión (45). Se han descrito éxitos similares para el análisis de variación del número de copias (CNV) (46,47).
Además de los paradigmas para la detección de variantes estándar, DeepVariant de Google transformó un problema de convencional en otro para el reconocimiento de imágenes al convertir un archivo BAM en imágenes similares a las instantáneas del navegador del genoma, donde el llamado de variantes se hace utilizando el marco Inception Tensor Flow que se desarrolló originalmente para la clasificación visual computarizada (48). Otro estudio reciente aplicó con éxito el machine learning para la secuenciación de datos de múltiples regiones de un tumor permitiendo identificar y aprender patrones de crecimiento como predictores precisos de la progresión del tumor (49). Adicionalmente, se están entrenando otros modelos de IA para caracterización de estructuras secundarias incluyendo fosforilación proteica en respuesta a la administración de medicamentos (contemplando la dosis biológica efectiva) (50). Finalmente, el proceso de depuración depende de la homologación de decisiones de la IA a partir de nociones clínicas con enorme variabilidad intra e interindividual (Figura 4). Para validar el papel de múltiples modelos de machine learning, el hospital Mash General Hospital diseñó un estudio que incluyó ~500 características clínicas y cerca de 20.000 variantes somáticas con potencial en la toma de decisiones. La comparación de la estructura de IA contra el Genomic Tumor Board demostró que el uso de una escala basada en regresión logística tuvo una tasa de falsos negativos y positivos del 1 y 2%, respectivamente, hallazgo que resultó comparable a las decisiones humanas (51).

Figura 4. Interacción dinámica desde la clínica (información relacional de la sintomatología, imágenes y características del paciente integradas en la historia clínica electrónica) hacia la genómica y bioinformática a través de algoritmos de análisis multicapa que permiten la subselección de intervenciones terapéuticas basadas en medicina de precisión.
Por otra parte, el volumen de literatura médica relacionado con genómica tumoral resulta inmanejable (~165.000/año). Esta dimensión podría llegar a ser manejable usando herramientas que contemplen el procesamiento natural del lenguaje para reducir el tiempo y esfuerzo necesarios para la recuperación de la información que permita la generación de nuevas hipótesis basadas en la mejor evidencia (Figura 5). La minería de datos también ha permitido el reconocimiento de entidades a través de proceso digitales de nominación 8Bio-NER) facilitando la extracción de referencias en medicina de precisión.
Desafortunadamente, no existe un estándar universal para denominar las variantes genéticas y existen múltiples formas de presentar el mismo evento en la literatura y en las bases de datos genómicas. Para consolidar el conocimiento sobre variantes patogénicas a partir de la literatura e integrarlas con los datos curados en recursos existentes como ClinVar y COSMIC, resulta esencial el uso correcto de la nomenclatura HGVS así como la introducción del número de identificación del SNP de referencia (RSID) (52). Recientemente, se han aplicado varios métodos de aprendizaje profundo al reconocimiento de entidades con nombre biomédico y sus respectivas alteraciones genéticas con una ganancia significativa en el rendimiento para integrar mejor las características multidimensionales y, al mismo tiempo, minimizar los requerimientos manuales (53).

Figura 5. Número de publicaciones frente al año de indexación. En esta figura se representan dos ejes “y”, uno exhibe el número de artículos relacionados con genómica tumoral, y el otro, el número de manuscritos asociados con genómica más NLP. El eje “x” representa el año de publicación. Tomado y modificado con autorización de Xu J, Yang P, Xue S, et al. Translating cancer genomics into precision medicine with artificial intelligence: applications, challenges and future perspectives. Hum Genet. 2019 Feb;138(2):109-124. doi: 10.1007/s00439-019-01970-5.
Retos para la implementación y uso de la IA en el ámbito de la genómica
La evaluación de la precisión de la IA con relación a la genómica es fundamental para titular el funcionamiento de los sistemas solventando el precepto de la “caja negra”. En la genómica tiene especial importancia la clasificación de variantes y su relevancia clínica, la validación de la literatura y la clasificación de vectores que permiten el diseño de biomarcadores. A pesar de la abundancia de información clínica y cruda de datos genómicos, la protección individual de estos documentos por las pautas HIPPA y GDPR limita el acceso a su estudio y uso para la capacitación y evaluación de los sistemas de IA aplicables al diseño de planes personalizados de tratamiento. En adición, la reproducibilidad de los resultados experimentales incluidos en los estudios de IA sigue siendo un problema para la implementación en la practica clínica regular. Debido a que los algoritmos de aprendizaje suelen tener múltiples componentes ajustables, el rendimiento suele verse afectado por la sensibilidad de la escala y calidad de los datos de entrenamiento, la configuración empírica de los parámetros, y los procesos de inicialización y optimización. Muchas publicaciones no revelan los supuestos simplificadores o los detalles de implementación y, por lo tanto, dificultan la reproducción de los resultados. En adición, la mayoría de los estudios no comparte el código fuente.
Conclusión
Si bien la salud digital se ha vuelto esencial para brindar las mejores prácticas en el cuidado sanitario, plantea desafíos sin precedentes para los pacientes, investigadores y para la comunidad biomédica, en especial, cuando confluye con la complejidad de la medicina de precisión y los análisis multiómicos. Por el momento, la intersección entre la IA y la genómica semeja a gigantes entre los gigantes, recordando la respuesta que algún día diera Isaac Asimov a la pregunta sobre el científico más grande de la historia. Después, de quedarse unos segundos en silencio replicó entre dientes “La historia probablemente aún no lo ha visto, sin embargo, tengo dudas sobre a quien colocar en segundo lugar”. Entonces, Asimov consideraba que para este lugar ya había una dura liza entre Albert Einstein, Ernest Rutherford, Niels Borh, Louis Pasteur, Charles Darwin, Galileo Galilei, Arquímedes y algunos otros. Lo que sí tenía claro, era que, al menos hasta donde su visión alcanzó, el mayor talento había sido de Isaac Newton. La IA transformará la historiografía de la biología molecular aplicada, en especial, para patologías complejas como el cáncer, donde la fuente del análisis avanzado de datos ya lo ha hecho y lo seguirá haciendo. Nada ha sido más estimulante que tener la oportunidad de vivirlo, nada vale más que reconocer que “antes pensábamos que nuestro futuro estaba en las estrellas, ahora sabemos que está en nuestros genes” (James Watson), y la IA esta al servicio de la curiosidad para leerlos.
LEER LA I PARTE. Caminando a hombros de gigantes: intersección entre la genómica y la IA. I Parte
___________
Andrés F. Cardona, MD MSc PhD MBA, Grupo Oncología Clínica y Traslacional, Clínica del Country, Bogotá, Colombia. Fundación para la Investigación Clínica y Molecular Aplicada del Cáncer – FICMAC, Bogotá, Colombia. Grupo de Investigación en Oncología Molecular y Sistemas Biológicos, (Fox-G), Universidad El Bosque, Bogotá, Colombia
Este artículo es republicado de AIpocrates. Para ver el artículo original en Aipocrates.org
Visitas: 18



