
En la revista Medicina No. 107 del año 2014, un grupo de investigadoras y para esa fecha estudiantes de Doctorado en Ciencias Biológicas de la Universidad de Los Andes, publicaron un artículo titulado “Genética molecular de la Hemofilia A en una familia colombiana con diagnóstico de enfermedad de Von Willebrand y de Hemofilia A”. Con motivo del Día Mundial de la Hemofilia, traemos apartes del artículo.
Por Diana Carolina Polanía Villanueva, Diana María Narváez Noguera, Helena Groot de Restrepo.
El Factor von Willebrand circula en el plasma formando un complejo con el Factor VIII de coagulación por enlaces no covalentes. Esta interacción evita la degradación enzimática del Factor VIII y asegura su transporte al lugar de formación del coágulo de fibrina. Debido a su estrecha relación, la disminución de la actividad de un factor puede afectar la actividad del otro, lo que genera un diagnóstico clínico equivocado en cuanto a qué enfermedad se padece, si Hemofilia A o Enfermedad de Von Willebrand.
Este estudio reporta el caso de una familia colombiana que según diagnóstico clínico de su fenotipo, padecía las dos enfermedades. Sin embargo, dicha familia carecía de un estudio genético que permitiera verificar y contrastar el diagnóstico que hacen las entidades de salud. Por tal razón, se realizó un diagnóstico genético por pruebas moleculares que detectan mutaciones, como las inversiones en los intrones 1 y 22 por PCR de fragmentos largos y la secuenciación del gen del Factor VIII, esta última no aplicada y publicada en Colombia hasta el momento.
Se encontraron dos mutaciones sinónimas en los exones 14 y 26 que no alteran la secuencia de aminoácidos en la proteína; por tanto, se descarta la presencia de Hemofilia A en la familia. Se plantea la posibilidad de un caso de Enfermedad de von Willebrand únicamente. El estudio demuestra la necesidad que hay en el país de ampliar las pruebas clínicas y de incluir el diagnóstico genético en casos de ambigüedad en el diagnóstico de estas coagulopatías.
INTRODUCCIÓN.
La Hemofilia A es un trastorno genético recesivo ligado al sexo, que da como resultado una deficiencia en la actividad coagulante del Factor VIII del plasma, siendo la frecuencia de incidencia de 1:5,000 y 1:10,000 varones nacidos vivos dependiendo de la población (1-3). La Enfermedad de von Willebrand; por su parte, es un trastorno hereditario autosómico en el que hay deficiencia o disfunción del Factor von Willebrand. La Enfermedad de von Willebrand afecta alrededor del 1% de la población mundial y se considera el trastorno de coagulación menos severo (4, 5).
El gen del Factor VIII se ubica en el brazo largo del cromosoma X. Comprende 186 kb, 26 exones y 25 intrones, algunos de ellos muy grandes como el intrón 22 (16424 pb). Este gen tiene dos copias intragénicas (Factor VIIIA y Factor VIIIB) dentro del intrón 22 y dos copias extragénicas en la región telomérica que son altamente homólogas con las intragénicas. El gen del Factor von Willebrand se ubica en el brazo corto del cromosoma 12 en la posición p13.3, abarca 178 kb y constituye 52 exones cuyo tamaño oscila entre 1.3kb (exón 28) y 40 pb (exón 50).
Tanto el Factor von Willebrand como el Factor VIII juegan un papel importante en la hemostasia para formar un retículo de fibras de fibrina estable en los coágulos de sangre. El Factor von Willebrand circula en el plasma, estabiliza y forma un complejo con el Factor VIII por enlaces no covalentes. Esta interacción evita la degradación enzimática del Factor VIII y asegura su transporte al lugar de la formación del coágulo de fibrina, en donde promueve la adhesión y agregación plaquetaria al subendotelio. Allí el Factor VIII es activado a Factor VIIIA y actúa como cofactor para el Factor IX activado, el cual promueve un paso más en la cascada de coagulación sanguínea hasta formar el coágulo.
En el último reporte de la Federación Mundial de la Hemofilia se identificaron 400.000 personas que viven con desórdenes de coagulación en 108 países. En Colombia se calculan aproximadamente unos 2.800 pacientes. En el 2008, por ejemplo, se reportaron en el país, 1.658 pacientes con Hemofilia, 1.255 con Hemofilia A, 272 con Hemofilia B y 131 con Hemofilias de tipo desconocido. Sólo 1.446 hemofílicos están registrados en la Liga Colombiana de Hemofilia, incluyendo 54 con desarrollo de inhibidores, 303 casos de Hemofilia B y 208 casos de Enfermedad de von Willebrand.
La Enfermedad de von Willebrand en particular, es una anomalía tanto en la cantidad como en la estructura del Factor von Willebrand y se traduce en una alteración de la función plaquetaria en lo que respecta a su adhesión para formar el tapón blando o trombo blanco. Aunque el Factor von Willebrand actúa como transportador y estabilizador del Factor VIII, su principal función es participar en la función plaquetaria, concretamente en la adhesión de las plaquetas a las fibras de colágeno que quedan al descubierto tras una herida o daño endotelial, hecho que inicia una cascada de interacciones entre las plaquetas, el Factor von Willebrand y las glucoproteínas Ib/IX y IIb/IIIa, hasta unirse al fibrinógeno, para mediar la agregación plaquetaria irreversible.
En la actualidad, la clasificación de la Enfermedad de von Willebrand se realiza con base en la concentración plasmática del Factor von Willebrand, a la actividad biológica y a los patrones de los multímeros. Factores como el sistema eritrocitario ABO del individuo pueden influir en la presentación y patología de la Enfermedad de von Willebrand. Aquellos individuos con grupo sanguíneo O tienen un nivel medio menor al de personas con otros grupos sanguíneos. A menos que el antígeno del Factor von Willebrand específico del grupo ABO referencie rangos habituales, los individuos del grupo O pueden ser diagnosticados erróneamente con Enfermedad de von Willebrand tipo 1. Algunas personas de grupo sanguíneo AB con un defecto genético del Factor von Willebrand pueden pasar por alto el diagnóstico porque los niveles son elevados debido a su grupo sanguíneo.
Cabe resaltar que desde el punto de vista del diagnóstico clínico las dos enfermedades son difícilmente distinguibles, pues las dos proteínas (Factor VIII y Factor von Willebrand) circulan juntas en la sangre y la disminución en la cantidad del Factor VIII también puede significar una disminución del Factor von Willebrand. Tanto la Enfermedad de von Willebrand como la Hemofilia A presentan heterogeneidad alélica y expresividad variable. La expresividad variable, por ejemplo, es analizada dependiendo de la cantidad de Factor VIII o Factor von Willebrand en la sangre. Resultados del nivel de los Factores menores al 1% se clasifican como graves o severos, entre 1 a 5% moderados y entre 5 a 49% leves para Factor VIII.
En el caso del Factor von Willebrand, entre 5 y 60% son considerados casos leves para grupos sanguíneos A, B y AB; y entre 5 – 40% casos leves para el grupo sanguíneo O. Adicionalmente, las dos enfermedades se describen como hemorragias o hematomas frecuentes o esporádicos sin causa aparente o ante un traumatismo en las superficies mucosas de nariz y boca. En casos severos, las hemorragias ocurren en las articulaciones mayores de las extremidades (rodillas, tobillos y codos) y, en menor grado, en caderas, hombros y músculos grandes. Con frecuencia, las mujeres con Enfermedad de von Willebrand presentan menorragia y abortos con hemorragias severas. Esto como consecuencia de que dicha enfermedad puede afectar por igual a hombres y mujeres, a diferencia de la Hemofilia, que en la mayoría de los casos sólo es padecida por hombres.
La sintomatología aparentemente similar puede llevar a dar diagnósticos poco acertados de la condición clínica de los pacientes. En algunos casos, incluso se puede llegar a diagnosticar Hemofilia, cuando quizás la enfermedad presente sea von Willebrand o viceversa. Adicionalmente, las entidades de salud hacen inferencias sobre el genotipo de los pacientes, atribuyéndoles el ser portadores o no de dichas enfermedades, sin corroborar los resultados de laboratorio con el correspondiente estudio genético. El trabajo estudió el primer reporte en Colombia de una familia con doble diagnóstico clínico de Hemofilia A y Enfermedad de von Willebrand y buscó comprobar genéticamente si dicho diagnóstico era cierto, basándose en el análisis molecular del Factor VIII.
MATERIALES Y MÉTODOS
Muestra de estudio.
La investigación tuvo como muestra una familia colombiana de tres generaciones entre los 6 y 65 años con doble diagnóstico clínico (Figura 1), independientemente de su sexo, edad, posibles comorbilidades u otras características sociodemográficas. Se excluyeron aquellos parientes que no presentaran vínculo consanguíneo. Según diagnóstico clínico de las diferentes Entidades Promotoras de Salud, la familia presentaba: un niño con Hemofilia A y Enfermedad de von Willebrand (Figura 1. Individuo III.1), una mujer que padece Enfermedad de von Willebrand (Figura 1. Individuo II.1) y una mujer portadora de Hemofilia A (Figura 1. Individuo II.2). Los otros dos integrantes de la familia no habían sido diagnosticados clínicamente. Los miembros de la familia que participaron en el proyecto firmaron un consentimiento informado en el que autorizaron la toma de muestras de sangre y el uso de su material genético en este estudio. El trabajo fue aprobado por el Comité de Ética de la Universidad de Los Andes, según Acta 211 de 2013.
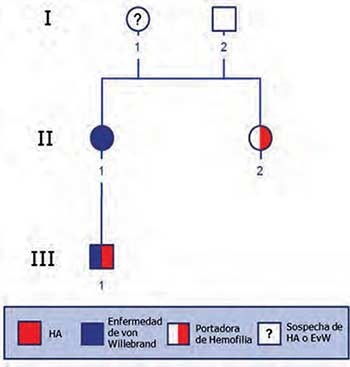
Genealogía de la familia objeto de estudio
Los resultados del estudio y el artículo completo para descargar en: Genética molecular de la Hemofilia A en una familia colombiana con diagnóstico de enfermedad de Von Willebrand y de Hemofilia A
LAS AUTORAS:
Diana Carolina Polanía Villanueva. Lic. en Biología, Magíster en Ciencias Biológicas área Biología, Ph.D. en Ciencias – Biológicas de la Universidad de Los Andes, Bogotá.
Diana María Narváez Noguera, Magister en Ciencias Biológicas, área Biología, Universidad de los Andes Doctora en Biología: Universidad de los Andes.
Helena Groot de Restrepo, Microbiol., MSc. Profesora Titular Facultad de Ciencias, Departamento de Ciencias Biológicas. Directora del Laboratorio de Genética Humana, Universidad de los Andes, Bogotá, Colombia.
Visitas: 99



