

José María Valpuesta, Centro Nacional de Biotecnología (CNB – CSIC)
La mayor parte de los procesos celulares son ejecutados por proteínas, y casi todas las enfermedades que sufrimos son producto de su mal funcionamiento. Resulta lógico, entonces, nuestro interés por entender cómo funcionan y por qué lo hacen mal, lo que ya adelanto que en la mayor parte de las ocasiones tiene que ver con mutaciones en la secuencia que dan lugar a cambios en su estructura.
Es, pues, muy importante conocer la estructura de las proteínas, y eso se puede conseguir mediante técnicas experimentales –difracción de rayos X, resonancia magnética nuclear y criomicroscopía electrónica– o, potencialmente, a través de técnicas de predicción, de las que el programa Alphafold es el mejor representante. Este poderoso sistema de inteligencia artificial es capaz de predeterminar, con mayor o menor exactitud, la forma tridimensional de 200 millones de proteínas. Prácticamente, según sus responsables, “todo el universo” de estas piezas fundamentales de la vida.
Todas están formadas por 20 aminoácidos
Para entender su importancia, tenemos que ir hacia atrás en el tiempo, a mediados del siglo XIX, cuando las proteínas fueron caracterizadas como una de las cinco grandes familias de moléculas biológicas –las otras son los ácidos nucleicos, los azúcares, los lípidos y un cajón de sastre donde se colocan ciertas hormonas–.
En las décadas siguientes se descubrió cuáles son los componentes básicos de las proteínas: los aminoácidos. Esa búsqueda determinó que son 20 los aminoácidos que fundamentalmente constituyen las proteínas de todos los organismos.
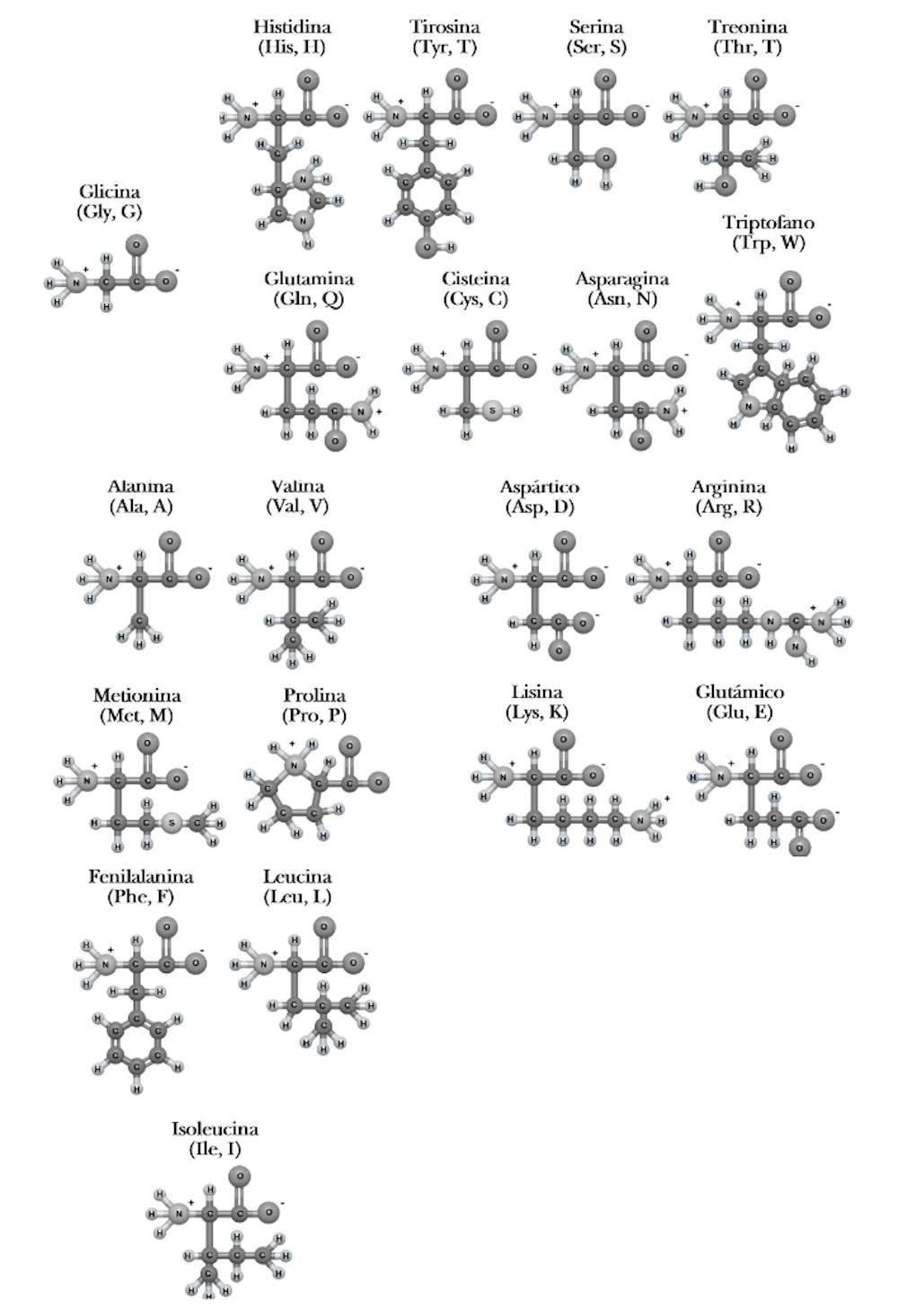
Los 20 aminoácidos ´canónicos’, cada uno con sus características únicas que contribuyen a la estructura de las proteínas.Author provided
Todos ellos tienen un componente común, que es un grupo químico cargado negativamente –un grupo carboxilo– en un extremo de la molécula, y otro cargado positivamente –un grupo amino– en el otro extremo. La reacción entre el grupo carboxilo de un aminoácido y el grupo amino de otro genera el llamado enlace peptídico, y la repetición de estas interacciones da a lugar a una estructura polimérica lineal, a un espagueti.
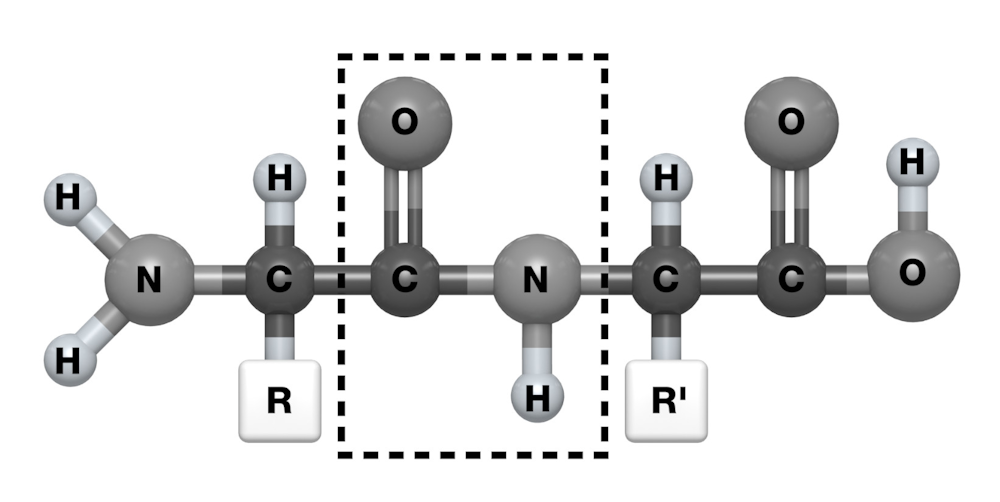
El enlace peptídico (rectángulo), que se produce entre dos aminoácidos contiguos en la secuencia.Author provided
Lo que hace diferente a los 20 aminoácidos es el resto de la molécula, conocida como cadena lateral. Esta puede ser más o menos grande, poseer cargas positivas o negativas y tener naturaleza hidrófila –le gusta interactuar con el agua que le rodea– o hidrófoba –intenta evitar interactuar con las moléculas de agua–.
La importancia de la estructura 3D
¿Por qué esas diferencias entre los aminoácidos? La respuesta está en los enlaces de naturaleza débil, que se descubrieron a principios del siglo XX. Estas ligaduras –puentes de hidrógeno, enlaces de van der Waals…– pueden establecerse entre átomos de la cadena lateral de un aminoácido con otras de aminoácidos lejanos. ¿Qué significa esto? Que a resultas de esas interacciones más o menos lejanas, el espagueti deja de ser lineal y comienza a plegarse en el espacio, a tener una estructura tridimensional.
Aproximadamente a mediados del siglo XX, dos líneas de trabajo convergieron. Una, utilizando enfoques bioquímicos, pudo demostrar que cada proteína –por ejemplo, la insulina– tiene una secuencia de aminoácidos única y distinta de las otras. La función está pues incluida de alguna manera en su secuencia lineal.
El segundo campo de investigación se interesó por la tridimensionalidad y se basó en el uso de la técnica de difracción de rayos X sobre cristales. Para conseguir cristales de proteínas –o de cualquier otra molécula–, debe producirse una disposición ordenada de ellas, y para ello es imprescindible que sean iguales. Esto dejaba claro ya en esa época que todas las moléculas de proteína en el cristal poseen una misma estructura.
Los estudios de difracción sobre cristales de proteínas mostraron en una primera instancia la presencia de elementos regulares en la estructura de estos componentes biológicos, que uno de los genios del siglo XX, Linus Pauling, interpretó correctamente y nombró como hélice α y lámina β. Estos dos tipos de estructuras, mantenidas por los enlaces débiles antes referidos, sirven de armazón estructural al conjunto de la proteína.
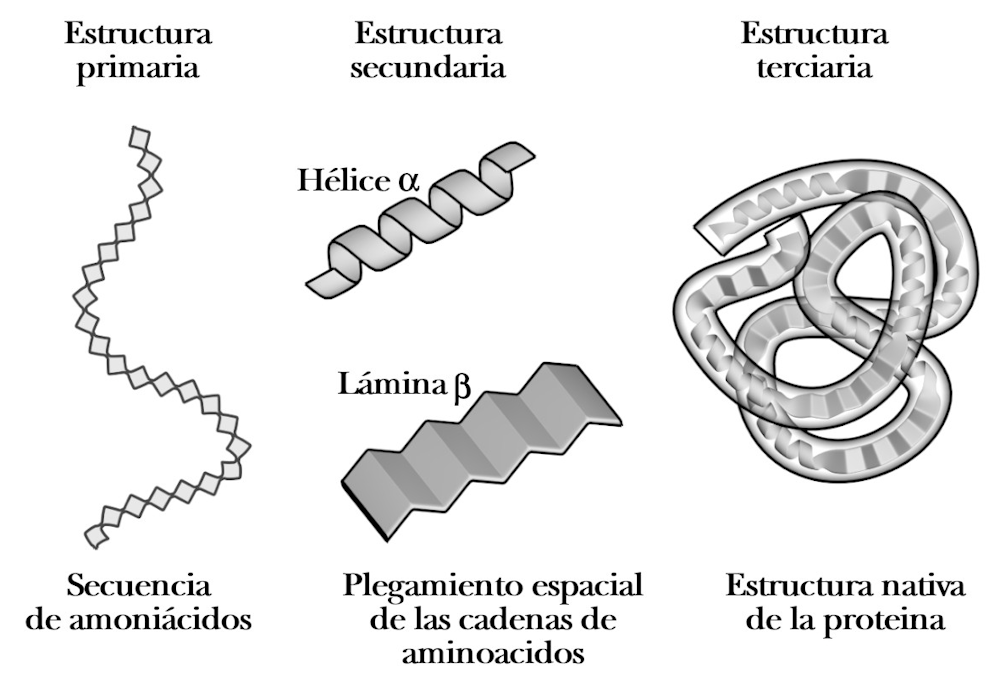
A la izquierda, la secuencia lineal de aminoácidos se llama estructura primaria; en el centro, en ciertas partes de la secuencia se producen estructuras ‘regulares’ que dan lugar a la estructura secundaria; a la derecha, el plegamiento general de toda la secuencia da lugar a la estructura terciaria (la estructura tridimensional).Author provided
A finales de los años cincuenta, el desarrollo de la técnica de difracción permitió observar las primeras estructuras proteicas a nivel atómico. Cuando en los siguientes años se determinaron las estructuras de moléculas homólogas –por ejemplo, la hemoglobina de caballo y la de cachalote– pudo comprobarse que las proteínas cuya función es la misma tienen además una secuencia muy similar que da lugar a una estructura casi idéntica.
El corolario de los descubrimientos llevados a cabo por esas dos líneas de investigación es que hay una relación directa entre la secuencia de aminoácidos de las proteínas, su estructura y su función.
Cómo predecir su forma tridimensional
Entonces, ¿si conocemos la secuencia de una proteína somos capaces de predecir su estructura tridimensional? No en todas las condiciones y no de una manera absolutamente fiable. Conocemos mucho de las propiedades físicas y químicas de los 20 aminoácidos y disponemos de herramientas poderosas para el cálculo de la conformación más estable de una determinada secuencia.
Todo esto nos ha permitido hacer predicciones muy fiables de lo que se conoce como estructura secundaria, la sucesión de hélices α, láminas β y zonas desordenadas a lo largo de la secuencia. Sin embargo, la determinación de cómo se pliegan esas hélices y láminas entre sí para dar la estructura tridimensional final es mucho más compleja.
Actualmente, la aparición de programas como el antes citado Alphafold está revolucionando esta área de investigación. Este y otros sistemas se basan en el conocimiento acumulado durante décadas en la base de datos de estructuras de proteínas (suma más de 150 000 en la actualidad). Utilizan conceptos que han estado en el campo durante décadas pero que, gracias al acceso a grandes cantidades de datos sobre distintos genomas y al uso de un tipo especial de algoritmos de análisis denominados de deep learning, han permitido dar un gran salto en el ámbito de la predicción tridimensional.
Este enfoque funciona muy bien cuando la proteína está bien representada en las bases de datos o cuando hay muchas secuencias similares que pueden ser alineadas entre sí, pero lo hace bastante peor cuando no es así o se encuentra formando complejos con otras proteínas. En este sentido, el problema de la predicción de la estructura tridimensional de las proteínas a partir del mero conocimiento de su secuencia de aminoácidos –el llamado problema del plegamiento– todavía no se ha resuelto.![]()
José María Valpuesta, Profesor de Investigación del CSIC. Jefe del Departamento de Estructura de Macromoléculas, Centro Nacional de Biotecnología (CNB – CSIC)
Este artículo fue publicado originalmente en The Conversation. Lea el original.